

Par
L’hypertension artérielle affaiblit presque toujours le cœur.
Étonnamment, certains patients porteurs du gène PDE3A muté étaient immunisés contre les dommages liés à l’hypertension.
Des scientifiques de Berlin étudient depuis des décennies une étrange maladie héréditaire qui fait que la moitié des personnes de certaines familles ont des doigts incroyablement courts et une pression artérielle anormalement élevée. S’ils ne sont pas traités, les individus atteints meurent souvent d’un accident vasculaire cérébral à l’âge de 50 ans. Des chercheurs du Max Delbrück Center (MDC) à Berlin ont découvert l’origine de la maladie en 2015 et ont pu la vérifier cinq ans plus tard à l’aide de modèles animaux : une mutation dans le gène de la phosphodiestérase 3A (PDE3A) rend son enzyme codée hyperactive, altérant la croissance osseuse et provoquant une hyperplasie des vaisseaux sanguins, entraînant une hypertension artérielle.
Immunisé contre les dommages liés à l’hypertension
“L’hypertension artérielle entraîne presque toujours un affaiblissement du cœur”, explique le Dr Enno Klußmann, responsable du Laboratoire de signalisation ancrée au Centre Max Delbrück et scientifique au Centre allemand de recherche cardiovasculaire (DZHK). Comme il doit pomper contre une pression plus élevée, explique Klußmann, l’organe essaie de renforcer son ventricule gauche. “Mais en fin de compte, cela entraîne l’épaississement du muscle cardiaque – connu sous le nom d’hypertrophie cardiaque – qui peut entraîner une insuffisance cardiaque diminuant considérablement sa capacité de pompage.”
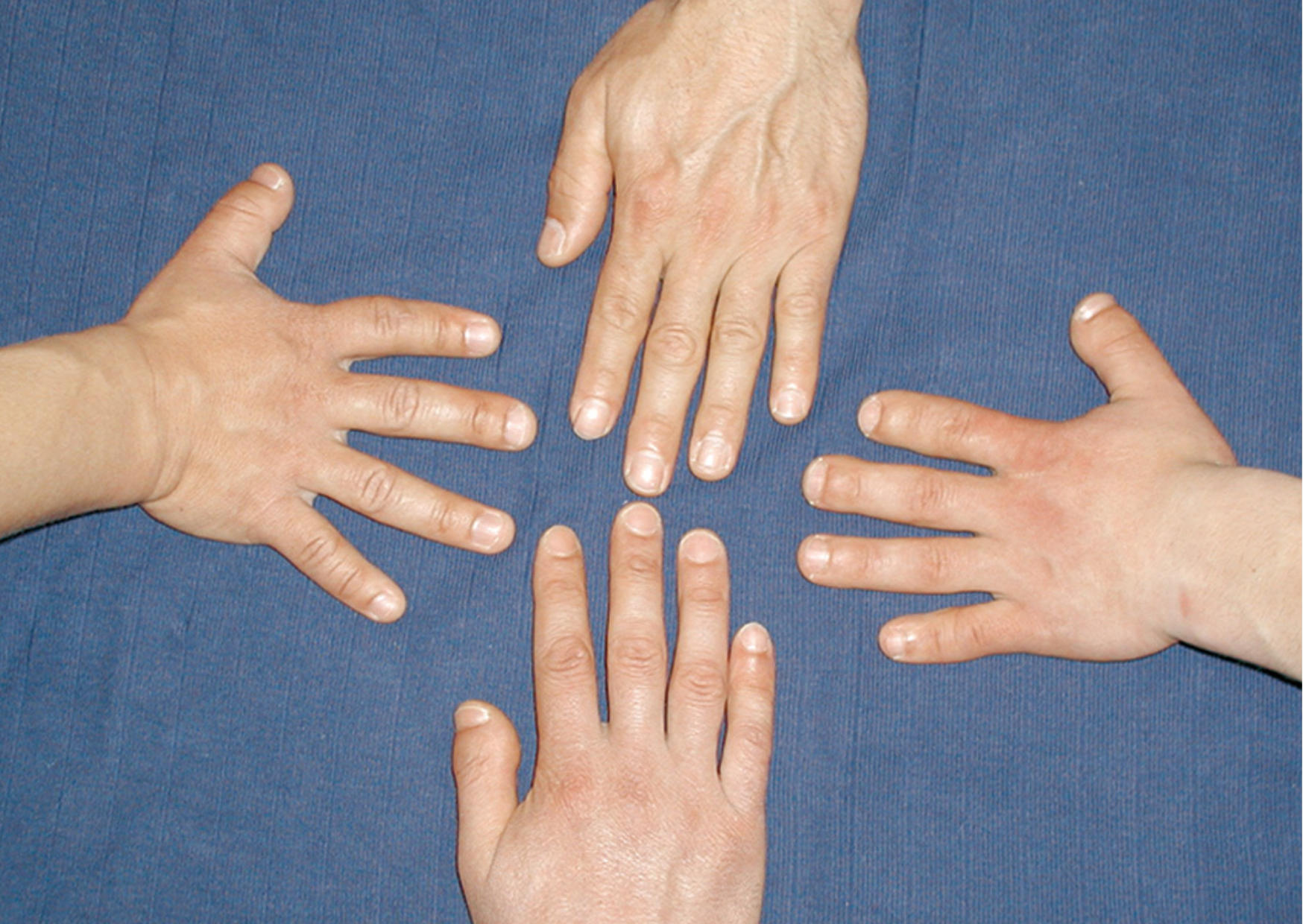
Doigts courts dans une famille. Crédit : Sylvia Bahring
Cependant, cela ne se produit pas chez les patients hypertendus avec des doigts courts et des gènes PDE3A mutants. “Pour des raisons qui sont maintenant en partie – mais pas encore entièrement – comprises, leur cœur semble immunisé contre les dommages qui résultent généralement de l’hypertension artérielle”, explique Klußmann.
La recherche a été menée par des scientifiques du Max Delbrück Center, de la Charité – Universitätsmedizin Berlin et du DZHK et a été publiée dans la revue Circulation. En plus de Klußmann, les auteurs finaux comprenaient les professeurs du Centre Max Delbrück Norbert Hübner et Michael Bader, ainsi que le Dr Sylvia Bähring du Centre de recherche expérimentale et clinique (ECRC), une institution conjointe de la Charité et du Centre Max Delbrück.
L’équipe, qui comprenait 43 autres chercheurs de Berlin, Bochum, Heidelberg, Cassel, Limbourg, Lübeck, Canada et Nouvelle-Zélande, a récemment publié ses découvertes sur les effets protecteurs de la mutation génétique – et pourquoi ces découvertes pourraient transformer la façon dont le cœur l’échec est traité à l’avenir. L’étude a quatre premiers auteurs, dont trois sont des chercheurs du Centre Max Delbrück et un de l’ECRC.
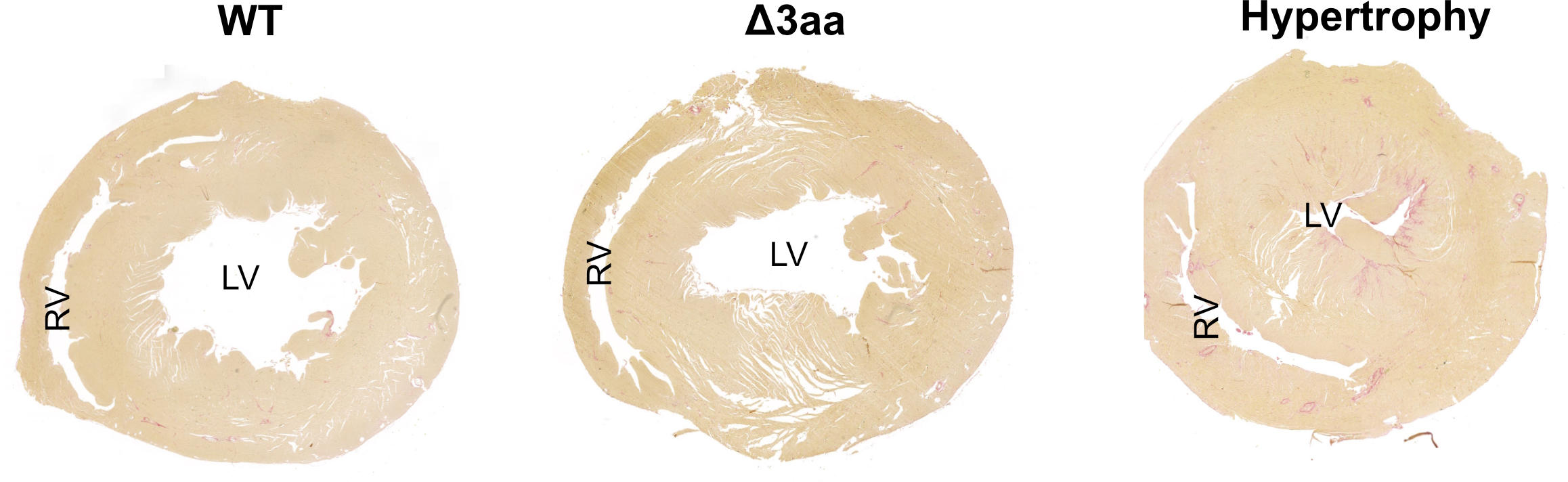
Coupe transversale à travers un cœur normal (à gauche), à travers l’un des cœurs mutants (au centre) et à travers un cœur sévèrement hypertrophique (à droite). Dans ce dernier, le ventricule gauche est agrandi. Crédit : Anastasiia Sholokh, MDC
Deux mutations avec le même effet
Les scientifiques ont effectué leurs tests sur des patients humains atteints du syndrome d’hypertension et de brachydactylie (HTNB), c’est-à-dire d’hypertension artérielle et de chiffres anormalement courts, ainsi que sur des modèles de rats et des cellules du muscle cardiaque. Les cellules ont été cultivées à partir de cellules souches spécialement conçues, appelées cellules souches pluripotentes induites. Avant le début des tests, les chercheurs ont modifié le gène PDE3A dans les cellules et les animaux pour imiter les mutations HTNB.
“Nous avons découvert une mutation du gène PDE3A jusque-là inconnue chez les patients que nous avons examinés”, rapporte Bähring. “Des études antérieures avaient toujours montré que la mutation de l’enzyme était située en dehors du domaine catalytique – mais nous avons maintenant trouvé une mutation en plein centre de ce domaine.” Étonnamment, les deux mutations ont le même effet en ce sens qu’elles rendent l’enzyme plus active que d’habitude. Cette hyperactivité accélère la dégradation de l’une des molécules de signalisation importantes de la cellule, connue sous le nom d’AMPc (adénosine monophosphate cyclique), qui est impliquée dans la contraction des cellules du muscle cardiaque. “Il est possible que cette modification génétique – quel que soit son emplacement – provoque le regroupement de deux ou plusieurs molécules PDE3A et donc leur efficacité”, soupçonne Bähring.
Les protéines restent les mêmes
Les chercheurs ont utilisé un modèle de rat – créé avec la technologie CRISPR-Cas9 par le laboratoire de Michael Bader au Max Delbrück Center – pour essayer de mieux comprendre les effets des mutations. “Nous avons traité les animaux avec l’agent isoprotérénol, un soi-disant agoniste des récepteurs bêta”, explique Klußmann. Ces médicaments sont parfois utilisés chez les patients atteints d’insuffisance cardiaque en phase terminale. L’isoprotérénol est connu pour induire une hypertrophie cardiaque. « Pourtant, étonnamment, cela s’est produit chez les rats génétiquement modifiés d’une manière similaire à ce que nous avons observé chez les animaux de type sauvage. Contrairement à ce à quoi nous nous attendions, l’hypertension existante n’a pas aggravé la situation », rapporte Klußmann. “Leurs cœurs étaient bien évidemment protégés de cet effet de l’isoprotérénol.”
Dans d’autres expériences, l’équipe a étudié si les protéines d’une cascade de signalisation spécifique des cellules du muscle cardiaque avaient changé à la suite de la mutation et, le cas échéant, lesquelles. Grâce à cette chaîne de réactions chimiques, le cœur réagit à l’adrénaline et bat plus vite en réponse à des situations telles que l’excitation. L’adrénaline active les récepteurs bêta des cellules, les obligeant à produire plus d’AMPc. La PDE3A et d’autres PDE arrêtent le processus en modifiant chimiquement l’AMPc. “Cependant, nous avons trouvé peu de différence entre les rats mutants et de type sauvage à la fois au niveau de la protéine et du
” data-gt-translate-attributes=”[{“attribute=””>ARN[{“attribute=””>RNA niveaux », déclare Klußmann.
Plus de calcium dans le cytosol
La conversion de l’AMPc par la PDE3A ne se produit pas n’importe où dans la cellule du muscle cardiaque, mais à proximité d’un système membranaire tubulaire qui stocke les ions calcium. Une libération de ces ions dans le cytosol de la cellule déclenche la contraction musculaire, faisant ainsi battre le cœur. Après la contraction, le calcium est pompé vers le stockage par un complexe protéique. Ce processus est également réglementé localement par PDE.
Klußmann et son équipe ont émis l’hypothèse que ces enzymes étant hyperactives dans la région locale autour de la pompe à calcium, il devrait y avoir moins d’AMPc, ce qui inhiberait l’activité de la pompe. “Dans les cellules du muscle cardiaque génétiquement modifiées, nous avons en fait montré que les ions calcium restent dans le cytosol plus longtemps que d’habitude”, explique le Dr Maria Ercu, membre du laboratoire de Klußmann et l’un des quatre premiers auteurs de l’étude. “Cela pourrait augmenter la force contractile des cellules.”
Activer au lieu d’inhiber
“Les inhibiteurs de la PDE3 sont actuellement utilisés pour le traitement de l’insuffisance cardiaque aiguë afin d’augmenter les niveaux d’AMPc”, explique Klußmann. Un traitement régulier avec ces médicaments saperait rapidement la force du muscle cardiaque. “Nos résultats suggèrent maintenant que non pas l’inhibition de la PDE3, mais – au contraire – l’activation sélective de la PDE3A peut être une approche nouvelle et considérablement améliorée pour prévenir et traiter les lésions cardiaques induites par l’hypertension comme la cardiomyopathie hypertrophique et l’insuffisance cardiaque”, déclare Klußmann. .
Mais avant que cela ne puisse se produire, dit-il, plus de lumière doit être faite sur les effets protecteurs de la mutation. “Nous avons observé que la PDE3A devient non seulement plus active, mais aussi que sa concentration dans les cellules du muscle cardiaque diminue”, rapporte le chercheur, ajoutant qu’il est possible que la première s’explique par l’oligomérisation – un mécanisme qui implique au moins deux enzymes. molécules travaillant ensemble. “Dans ce cas”, explique Klußmann, “nous pourrions probablement développer des stratégies qui initient artificiellement l’oligomérisation locale – imitant ainsi l’effet protecteur pour le cœur”.
Référence : « Mutant Phosphodiesterase 3A Protects From Hypertension-Induced Cardiac Damage » par Maria Ercu, Michael B. Mücke, Tamara Pallien, Lajos Markó, Anastasiia Sholokh, Carolin Schächterle, Atakan Aydin, Alexa Kidd, Stephan Walter, Yasmin Esmati, Brandon J. McMurray, Daniella F. Lato, Daniele Yumi Sunaga-Franze, Philip H. Dierks, Barbara Isabel Montesinos Flores, Ryan Walker-Gray, Maolian Gong, Claudia Merticariu, Kerstin Zühlke, Michael Russwurm, Tianan Liu, Theda UP Batolomaeus, Sabine Pautz, Stefanie Schelenz, Martin Taube, Hanna Napieczynska, Arnd Heuser, Jenny Eichhorst, Martin Lehmann, Duncan C. Miller, Sebastian Diecke, Fatimunnisa Qadri, Elena Popova, Reika Langanki, Matthew A. Movsesian, Friedrich W. Herberg, Sofia K. Forslund, Dominik N. Müller, Tatiana Borodina, Philipp G. Maass, Sylvia Bähring, Norbert Hübner, Michael Bader et Enno Klussmann, 19 octobre 2022, Circulation.
DOI : 10.1161/CIRCULATIONAHA.122.060210


:max_bytes(150000):strip_icc()/Health-GettyImages-859701970-ec460822b55a41ed8beed55dc960b3af.jpg?fit=300%2C300&ssl=1)
